














































Important notice
Please note that the images, figures, and tables for this Review have not been added yet. We are actively working to digitise and include these materials from our past magazines.
Main content
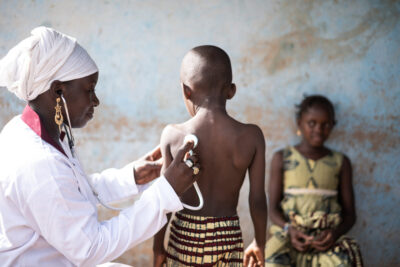
Sickle-cell disease (SCD) is an autosomal, recessive haemoglobinopathy and multisystem disorder characterised by episodes of vaso-occlusion, ongoing haemolytic anaemia and progressive organ failure. It is the most common monogenetic disease worldwide with an estimated 350.000 births annually affected and recognised as a global public health problem by the World Health Organization (WHO).[1] The global distribution of the sickle haemoglobin (HbS) allele is indicative of the protective effect of sickle cell trait (HbAS) against Plasmodium falciparum malaria, first described by Haldane in 1949.[2,3,4] However, migration from malaria-endemic regions to Northern America, Western Europe and Australia has led to spreading of the HbS allele far beyond its origin.[5] Roughly 2500 individuals in the Netherlands currently have SCD, of which 1000 are children, and the carrier incidence is 0.4%.[6] Most of those affected are of Surinamese, Asian or African ancestry, with a minority being of Afro-Caribbean or Middle Eastern descent.[7]
Seventy five percent of the global burden of SCD occurs in sub-Saharan Africa, where the majority of children with the disease do not reach their fifth birthday.[1,8] In contrast, the life expectancy in well-resourced countries has significantly improved with almost all infants now expected to survive into adulthood because of comprehensive care programs.[9,10] However, the average life expectancy of patients with SCD is still 20 years less than that of healthy adults.[11]
This article aims to provide insight into the characteristics of routine comprehensive care for patients with SCD in the Netherlands, with a special focus on paediatric aspects. It does not, however, provide a summary of the management of SCD, which is excellently reported in the guideline of the National Heart Lung and Blood Institute (NHLBI).[12]
Neonatal screening
Prompt diagnosis is the first step in improving treatment outcomes of individuals with SCD. Early diagnosis, before clinical symptoms occur, allows for the enrolment in comprehensive care programs and education of parents on the recognition of danger signs and importance of vaccinations and prophylactic antibiotics.
Increased migration from sickle-cell endemic countries to the Netherlands resulted in initiation of a universal newborn screening program for SCD in the Netherlands on 1 January 2007.[13] Diagnosis is established by high-performance liquid chromatography (HPLC), which has a high sensitivity and specificity and a positive predictive value of 100%. In addition, only a small blood sample is required and other inherited blood disorders such as severe α- and β- thalassemia are concomitantly diagnosed by this method.[14]
When a carrier of HbS is identified, both parents and the general practitioner (GP) receive a letter explaining the test results, except for parents who have indicated that they do not want to be informed about the carrier status of their child (opting out). Parents are then invited for an informative consultation about SCD and carriership. GPs are advised to emphasise the importance of testing both parents for haemoglobinopathies and to refer couples at-risk to a clinical geneticist. Knowledge of this risk allows for a range of options, including prenatal diagnosis and limiting of family size.
Vaccination and anti-biotic prophylaxis
Children with SCD are particularly at increased risk of severe bacterial infections due to an impaired splenic function. Prior to the initiation of neonatal screening for SCD, infection by encapsulated organisms was the leading cause of death in afflicted children.[16] In addition to routine courses of immunisations according to national schedules, yearly vaccinations against influenza from the age of 6 months are administered as well as repetitive pneumococcal vaccinations. Furthermore, all SCD patients receive twice-daily prophylactic penicillin from the age of 4 months until their twelfth birthday.
However, one-third of the children newly diagnosed with SCD are immigrants and not born in the Netherlands.[5] Unfortunately, those children are not systematically screened for haemoglobinopathies and are therefore at high risk of life-threating complications before the diagnosis SCD has been made. This is a disparity in healthcare and therefore the initiation of other screening programs is currently being considered (i.e., in health screening package offered to immigrants).
Specialised sickle- cell clinics
It is recommended that all patients diagnosed with SCD are referred to a (paediatric) haematologist with expertise in haemoglobinopathies to ensure good quality of care. Comprehensive medical care for SCD has significantly decreased morbidity and prolongs life expectancy for patients.[16] The Netherlands has specialised (paediatric) haematologists in all academic centres. Those centres are equipped with a dedicated sickle-cell care team consisting of a (paediatric) haematologist, sickle-cell nurse, clinical geneticist, social worker and psychologist, working closely with a variety of sub-specialisms with expertise in SCD (e.g. neurology, cardiology, pulmonology, nephrology, ophthalmology, orthopaedics). This multidisciplinary approach allows for comprehensive, family-centred care for children and adults with SCD. Unfortunately, an extensive analysis of Dutch paediatric SCD patients showed that not all children are provided with such care.[6] This could be due to insufficient access to the Dutch healthcare system by minorities or potential unawareness among health professionals of the importance of comprehensive care for ‘less severe’ SCD. Regarding the latter, it is of the utmost importance to identify patients and to establish collaborations between paediatricians and haematologists with less expertise on the one hand and specialists in comprehensive care centres on the other, in order to provide the best care possible given current scientific knowledge.
Multiple sickle cells in a blood smear
Comprehensive medical evaluations
In summary, all paediatric and adult SCD patients need at least half-yearly scheduled medical evaluations to document baseline physical findings, including blood pressure and peripheral oxygen saturation and the evaluation of growth and development, as well as yearly sampling of blood and urine to detect organ failure. In addition, patients are monitored for complications of SCD including transcranial Doppler (TCD) ultrasonography (due to the high incidence of cerebral infarction [17]) and dilated eye examination (to screen for proliferative retinopathy). If the course of disease is severe or symptoms of deterioration or organ failure occur, treatment is intensified (hydroxyurea, blood- or exchange transfusions) and patients are immediately referred to sub-specialists with SCD expertise. Furthermore, patients and their parents are also followed with regard to quality of life by using validated questionnaires. The results are then integrated in clinical practice to address psychosocial issues in an efficient and effective manner.
Sickle cell outcome research (score)
The Dutch SCORE consortium was founded in 2016 and is composed of (paediatric) haematologists from seven comprehensive sickle-cell centres (Amsterdam, Rotterdam, Leiden, The Hague, Utrecht, Nijmegen and Groningen). SCORE is designed as a multicentre, prospective cohort study which aims to include the majority of SCD patients in the Netherlands. The goal of the collaboration is to identify factors and biomarkers contributing to the morbidity and mortality of SCD by uniformly collecting data. The research focuses on combining doctor-reported outcome measures (measures assessed by physicians, also called clinical data) with outcome measures from the patients’ perspective, also called PROMs (patient reported outcome measures) and PREMs (patient reported experience measures). By incorporating the patients’ and families’ values, beliefs and cultural norms, the impact of treatment and care can be fully evaluated.
Conclusion
Comprehensive care for patients with SCD is a time-intensive endeavour that includes neonatal screening, vaccination and antibiotic prophylaxis, periodic evaluations with screening for complications, and ongoing education of patients and relatives in specialised sickle-cell centres. Nevertheless, even with the best care, many SCD patients still face a lifetime of complications. Methodologically sound research is needed to address evidence gaps and improve our understanding of the best care for these patients. The SCORE initiative hopes to unravel the heterogeneity of the disease and to assess the impact of SCD on the wellbeing of patients and their families.
CORRESPONDING AUTHOR: M.HOUWING@ERASMUSMC.NL
References
- Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010-2050: modelling based on demographics, excess mortality, and interventions. PLOS Medicine 2013;10:7.
- Allison AC. Protection Afforded by Sickle-cell Trait Against Subtertian Malarial Infection. BMJ 1954;1:290-4.
- May J, Evans JA, Timmann C, Ehmen C, Busch W, Thye T, Aqbenyega T, Horstmann RD. Hemoglobin variants and disease manifestations in severe falciparum malaria. JAMA 2007;297:20.
- Haldane, JBS. Disease and evolution. Supplement to La Ricerca Scientifica 1949;19.
- Piel FB, Tatem AJ, Huang Z, Gupta S, Williams TN, Weatherall DJ. Global migration and the changing distribution of sickle haemoglobin: a quantitative study of temporal trends between 1960 and 2000. Lancet 2014;2:2.
- Peters M, Fijnvandraat CJ, van den Tweel XW, Garre FG, Giordano PC, van Wouwe JP, Rodriques Pereira R, Verkerk PH. One-third of the new paediatric patients with sickle cell disease in The Netherlands are immigrants and do not benefit from neonatal screening. Arch Dis Child 2010;95:10.
- Suijker MH, Roovers EA, Fijnvandraat CJ, Dors N, Rodriques Pereira R, Giordano PC, Verkerk PH, Peters M. Haemoglobinopathy in the 21st century: incidence, diagnosis and heel prick screening. Ned Tijdschr Geneeskd 2014;158: A7365.
- McGann PT, Hernandez AG, Ware RE. Sickle cell anemia in sub-Saharan Africa: advancing the clinical paradigm through partnerships and research. Blood 2017;129:2.
- Quin CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood 2010;115:17.
- Van der Plas EM, Van den Tweel XW, Geskus RB, Heijboer H, Biemond BJ, Peters M, Fijnvandraat CJ. Mortality and causes of death in children with sickle cell disease in the Netherlands, before the introduction of neonatal screening. Br J Haematol 2011;155:1.
- Gardner K, Douiri A, Drasar E, Allman M, Mwiriqi A, Awoqbade M, Thein SL. Surviv al in adults with sickle cell disease in a high-income setting. Blood 2016;128:10.
- Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, Jordan L, Lanzkron SM, Lottenberg R, Savage WJ, Tanabe PJ, Ware RE, Murad MH, Goldsmith JC, Ortiz E, Fulwood R, Horton A, John-Sowah J. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:10.
- Bolhuis PA, Page-Christiaens GC. The advisory report ‘Neonatal screening’ from the Health Council of the Netherlands. Ned Tijdschr Geneeskd 2005;149:2857-60.
- Giordano PC. Starting neonatal screening for haemoglobinopathies in The Netherlands. J Clin Pathol 2009;62:18-21.
- Onwubalili JK. Sickle-cell anaemia: an explanation for the ancient myth of reincarnation in Nigeria. Lancet 1983;27:2.
- Vichinsky EP. Comprehensive care in sickle cell disease: its impact on morbidity and mortality. Sem Hematol 1991;28:3.
- Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S, Moohr JW, Wethers DL, Pegelow CH, Gill FM. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 1998; 91:1.


